Prader - Willi Syndrome

Prader-Willi Syndrome was first discovered in 1956 by five Swiss doctors; Andrea Prader, Alexis Labhart, Andrew Ziegler, Guido Fanconi and Heinrich Willi. Nine children were studied with similar characteristics including hypotonia at birth, intellectual disability, low muscle tone, short stature, incomplete sexual development, cognitive disabilities, problem behaviors, and a chronic feeling of hunger that can lead to excessive eating and life threatening obesity. Individuals with Prader-Willi cannot feel full and in many cases individuals who have been left unsupervised in the presence of food have eaten so much that their stomachs can explode causing serious injury or death. Prader-Willi is an extremely rare genetic disorder and occurs only in 1 in every 10,000-30,000 individuals. It is caused by a deletion or partial-deletion of the long arm (q) of their father's 15th chromosome. While research has been inconsistent regarding comparative IQ levels, most physicians suggest that the deletion of chromosome 15q causes lower IQ. There is no cure to Prader-Willi syndrome, but some hormone treatments such as a growth hormone, can ease some of the symptoms, while medical professionals can support the families of individuals with Prader-Willi by educating them on ways to support positive behaviors; more specifically behaviors around over-eating. Adults with Prader-Willi Syndrome can lead semi independent lives, case dependent, and with monitoring of their obesity, can live normal life spans.
Symptoms and Signs of Prader-Willi
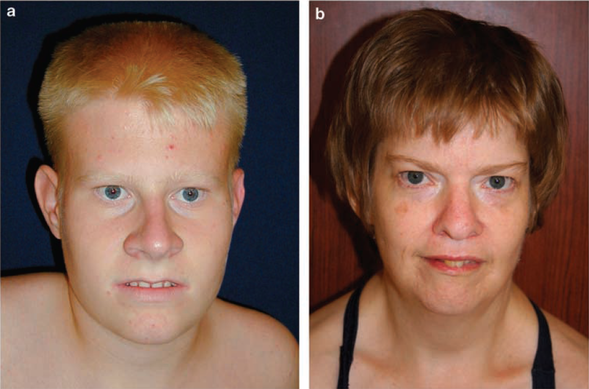
Prader-Willi California Foundation photograph of similar features found in Prader-Willi Syndrome patients. Subject A. is a 15 year old male and subject B. is a 41 year-old woman.
Uterus and birth
- Reduced fetal movement
- Frequent abnormal fetal position
- Occasional polyhydraminos(excessive amniotic fluid)
- Often breech or caesarean births
- Lethargy
- Hypotonia
- Feeding difficulties (due to poor muscle tone affecting sucking reflex)
- Difficulties establishing respiration
- Hypogonadism
- Delayed milestones/intellectual delay
- Excessive Sleeping
- Strabismus ('crossed eyes')
- Scoliosis (often not detected at birth)
- Cryptorchidism
- Speech Delay
- Poor physical coordination
- Hyperphagia over-eating) begins between the age of 2 and 8, and continues on throughout adulthood. Note change from feeding difficulties in infancy.
- Excessive Weight Gain
- Sleep Disorders
- Delayed puberty
- Short stature
- Obesity
- Extreme flexibility
- Infertility (males and females)
- High Threshold for Pain
- Hypogonadism
- Sparse pubic hair
- Obesity
- Hypotonia (low muscle tone)
- Learning disabilities/borderline intellectual functioning (but some cases of average intelligence)
- Prone to diabetes mellitus
- Extreme flexibility
- Prominent nasal bridge
- Small hands and feet with tapering of fingers
- Soft skin, which is easily bruised
- Excess fat, especially in the central portion of the body
- High, narrow forehead
- Thin upper lip
- Downturned mouth
- Almond-shaped eyes
- Light skin and hair relative to other family members
- Lack of complete sexual development
- Frequent skin picking
- Striae
- Delayed motor development
Diagnosis
A suspected diagnosis of Prader-Willi syndrome (PWS) is usually made by a physician based on clinical symptoms. PWS should be suspected in any infant born with significant hypotonia (muscle weakness or “floppiness”).If a patient meets 8 or more of the criterium for PWS they are recommended for genetic testing. The diagnosis is confirmed by a blood test. The preferred method of testing is a “methylation analysis,” which detects >99% of cases, including all of the major genetic subtypes of PWS (deletion, uniparental disomy, or imprinting mutation). A “FISH” (fluorescent in-situ hybridization) test will identify those patients with PWS due to a deletion, but it will not identify those who have Prader-Willi syndrome by “UPD” (uniparental disomy) or an imprinting error.
Almost all cases of PWS can be confirmed by one of the above tests. However, in the rare event that laboratory tests do not confirm PWS, a clinical diagnosis can be helpful for the development of a management plan.
Almost all cases of PWS can be confirmed by one of the above tests. However, in the rare event that laboratory tests do not confirm PWS, a clinical diagnosis can be helpful for the development of a management plan.
Management and Support Skills

|

|

|
Literary References for Management of PWS from left to right, Prader-Willi Syndrome: Coping with the Disease - Living with Those Involved by: Urs Eiholzer, Insatiable: A Prader-Willi Story by: Debbie Frisk and Chelsea McCutchin, Management of Prader-Willi Syndrome by: Merlin G. Butler, Phillip D.K. Lee, and Barbara Y. Whitman
As there is no cure for PWS, the only aid to curb symptoms such as picking and insatiable hunger is behavioral management. The most detailed, effective, and up-to-date supportive management suggestions are detailed by the Prader-Willi California Foundation or PWCF. The link for multiple resources as to the behavior management of both adults and children with Prader-Willi Syndrome are below.
PWS Case Study: Ryan
Meet Ryan:
Ryan is a 39 male diagnosed with PWS, Bi-Polar Disorder and Obsessive Compulsive Disorder. He currently attends a Day Program and works on the site of the Day Program as the site's "shredder" earning $9 an hour. He lives in a 24 hour care group home with two male roommates; one also diagnosed with Prader-Willi Syndrome, the other struggling with compulsive eating disorder. He remains under the guardianship of his parents and goes home every other weekend. He has been educated through Cardinal Cushing Centers throughout his adolescence and remains in their care throughout his adult life. Ryan is on a strictly regulated diet and earns a sparkling water at two different intervals throughout the day if he has been compliant with staff requests, does not display any outbursts or violence, and does not steal others food. Ryan frequently struggles with picking and also earns 3 altoids at two different intervals in the day for listening to staff when they cue him to stop picking. Ryan's staff in his residential program include a house manager as well as three rotating staff members who monitor him constantly to deter Ryan from picking. At his Day Program Ryan's ISP regulations are given by a behavior specialist, a DDS coordinator and a case manager; and his actions are monitored constantly by a 1:1 Employment Coach (Me). Ryan is not allowed to handle food at any time in his residential or day program except during meal or snack times. The fridges and cabinets are padlocked in both programs. Ryan loves to read but he struggles with comprehension. He also struggles with mathematics and critical thinking without staff prompting. He has a number of behaviors, one being excess question asking, to get staff or a peer's attention. When prompted he can answer the question himself 9 times out of 10. Ryan also has "pseudo-seizures" which have lasted periods up to 12 hours. These seizures were determined by several doctors to be solely behavioral and when cued that there is food in the room, Ryan immediately "wakes up". Despite the commonality that PWS causes individuals who suffer with it to be uncaring and uninterested in others, Ryan completely defies that statistic and cares about his peers immensely. Please refer to the documents below for additional information about Ryan.
Video Interviews
Works Cited
"Smith", Ryan (2015, July 25), an adult with Prader-Willi syndrome. (S.Curtis, Interviewer)
Eiholzer, Urs. Prader-Willi Syndrome Coping with the Disease, Living with Those Involved. Basel: Karger, 2005. Print.
Butler, Merlin Gene. Management of Prader-Willi Syndrome. 3rd ed. New York: Springer-Verlag, 2006. Print.
"Prader-Willi California Foundation | Our Vision: A Full Life Without Limits." PraderWilli California Foundation. Web. 3 Aug. 2015.
Frisk, Debbie. Insatiable: A Prader-Willi Story. Print.
Boer, H., Holland, A., Whittington, J., Butler, J., Webb, T., & Clarke, D. (2002). Psychotic illness in people with Prader-Willi syndrome due to chromosome 15 maternal uniparental disomy. The Lancet , 359, 135-136.
Burman, P., Ritzen, E., & Lindgren, A. (2001). Endocrine dysfunction in Prader-Willi syndrome: A review with special reference to GH. Endocrine Reviews , 22 (6), 787-799.
Cassidy, S., & Driscoll, D. (2009). Prader-Willi Syndrome. European Journal of Human Genetics (17), 3-13.
Cassidy, S., Forsythe, M., Heeger, S., Nicholls, R., Schork, N., Benn, P., et al. (1997). Comparison of phenotype between patients with Prader-Willi syndrome due to deletion 15q and uniparental disomy 15. American Journal of Medicinal Genetics (68), 433-440.
Cassidy, S., Schwartz, S., Miller, J., & Driscoll, D. (2012). Prader-Willi syndrome. Genetics in Medicine , 14 (4), 10-23.
Dykens, E., Cassidy, S., & King, B. (1999). Maladaptive behavior differences in Prader-Willi syndrome due to paternal deletion versus maternal uniparental disomy. American Journal on Mental Retardation , 104 (1), 67-77.
Dykens, E., Leckman, J., & Cassidy, S. (1996). Obsessions and Compulsions in Prader-Willi syndrome. Journal of Child Psychology and Psychiatry , 37 (8), 995-1002.
Fridman, C., Kok, F., & Koiffmann, C. (2000). Hypotonic infants and the Prader-Willi syndrome. Jornal de Pediatra , 76 (3), 246-250.
Holm, V., Cassidy, S., Butler, M., Hanchett, J., Greenswag, L., Whitman, B., et al. (1993). Prader-Willi syndrome: consensus diagnostic criteria. Pediatrics , 91 (2), 398-401.
Sinnema, M., Boer, H., Collin, P., Maaskant, M., Roozendall, K., Schrander-Stumpel, C., et al. (2011). Psychiatric Illness in a cohort of adults with Prader-Willi syndrome. Research in Developmental Disabilities (32), 1729-1735.
Staff, M. C. (2014, December 5). Angelman Syndrome. Retrieved July 19, 2015, from Mayoclinic.org: http://www.mayoclinic.org/diseases-conditions/angelman-syndrome/basics/definition/con-20033404
Staff, M. C. (2014, April 17). Prader-Willi syndrome. Retrieved July 10-20, 2015, from Mayoclinic.org: http://www.mayoclinic.org/diseases-conditions/prader-willi-syndrome/basics/complications/con-20028982
Swaab, D. (1997). Prader-Willi syndrome and the hypothalamus. Acta Paediatr , 50-54.
Veltman, M., Thompson, R., Roberts, S., Thomas, N., Whittington, J., & Bolton, P. (2004). Prader-Willi syndrome: A study comparing deletion and uniparental disomy cases with reference to autism spectrum disorders. European Child and Adolescent Psychiatry (13), 42-50.
Eiholzer, Urs. Prader-Willi Syndrome Coping with the Disease, Living with Those Involved. Basel: Karger, 2005. Print.
Butler, Merlin Gene. Management of Prader-Willi Syndrome. 3rd ed. New York: Springer-Verlag, 2006. Print.
"Prader-Willi California Foundation | Our Vision: A Full Life Without Limits." PraderWilli California Foundation. Web. 3 Aug. 2015.
Frisk, Debbie. Insatiable: A Prader-Willi Story. Print.
Boer, H., Holland, A., Whittington, J., Butler, J., Webb, T., & Clarke, D. (2002). Psychotic illness in people with Prader-Willi syndrome due to chromosome 15 maternal uniparental disomy. The Lancet , 359, 135-136.
Burman, P., Ritzen, E., & Lindgren, A. (2001). Endocrine dysfunction in Prader-Willi syndrome: A review with special reference to GH. Endocrine Reviews , 22 (6), 787-799.
Cassidy, S., & Driscoll, D. (2009). Prader-Willi Syndrome. European Journal of Human Genetics (17), 3-13.
Cassidy, S., Forsythe, M., Heeger, S., Nicholls, R., Schork, N., Benn, P., et al. (1997). Comparison of phenotype between patients with Prader-Willi syndrome due to deletion 15q and uniparental disomy 15. American Journal of Medicinal Genetics (68), 433-440.
Cassidy, S., Schwartz, S., Miller, J., & Driscoll, D. (2012). Prader-Willi syndrome. Genetics in Medicine , 14 (4), 10-23.
Dykens, E., Cassidy, S., & King, B. (1999). Maladaptive behavior differences in Prader-Willi syndrome due to paternal deletion versus maternal uniparental disomy. American Journal on Mental Retardation , 104 (1), 67-77.
Dykens, E., Leckman, J., & Cassidy, S. (1996). Obsessions and Compulsions in Prader-Willi syndrome. Journal of Child Psychology and Psychiatry , 37 (8), 995-1002.
Fridman, C., Kok, F., & Koiffmann, C. (2000). Hypotonic infants and the Prader-Willi syndrome. Jornal de Pediatra , 76 (3), 246-250.
Holm, V., Cassidy, S., Butler, M., Hanchett, J., Greenswag, L., Whitman, B., et al. (1993). Prader-Willi syndrome: consensus diagnostic criteria. Pediatrics , 91 (2), 398-401.
Sinnema, M., Boer, H., Collin, P., Maaskant, M., Roozendall, K., Schrander-Stumpel, C., et al. (2011). Psychiatric Illness in a cohort of adults with Prader-Willi syndrome. Research in Developmental Disabilities (32), 1729-1735.
Staff, M. C. (2014, December 5). Angelman Syndrome. Retrieved July 19, 2015, from Mayoclinic.org: http://www.mayoclinic.org/diseases-conditions/angelman-syndrome/basics/definition/con-20033404
Staff, M. C. (2014, April 17). Prader-Willi syndrome. Retrieved July 10-20, 2015, from Mayoclinic.org: http://www.mayoclinic.org/diseases-conditions/prader-willi-syndrome/basics/complications/con-20028982
Swaab, D. (1997). Prader-Willi syndrome and the hypothalamus. Acta Paediatr , 50-54.
Veltman, M., Thompson, R., Roberts, S., Thomas, N., Whittington, J., & Bolton, P. (2004). Prader-Willi syndrome: A study comparing deletion and uniparental disomy cases with reference to autism spectrum disorders. European Child and Adolescent Psychiatry (13), 42-50.